| Type: | Package |
| Title: | 'REST' 'API' Client for the 'NHGRI'-'EBI' 'GWAS' Catalog |
| Version: | 0.99.18 |
| Description: | 'GWAS' R 'API' Data Download. This package provides easy access to the 'NHGRI'-'EBI' 'GWAS' Catalog data by accessing the 'REST' 'API' https://www.ebi.ac.uk/gwas/rest/docs/api/. |
| Depends: | R (≥ 3.2.3) |
| License: | MIT + file LICENSE |
| URL: | https://github.com/ramiromagno/gwasrapidd, https://rmagno.eu/gwasrapidd/ |
| BugReports: | https://github.com/ramiromagno/gwasrapidd/issues |
| Encoding: | UTF-8 |
| Language: | en-US |
| LazyData: | true |
| RoxygenNote: | 7.3.2 |
| Config/Needs/website: | patterninstitute/chic |
| Imports: | magrittr, httr, urltools, pingr, stringr, dplyr, jsonlite, purrr, tibble, glue, tidyr (> 0.8.99), assertthat, rlang, methods, lubridate, plyr, testthat, utils, progress, writexl |
| Suggests: | httptest, spelling, knitr, rmarkdown, bookdown |
| Collate: | 'browser.R' 'cc.R' 'class-associations.R' 'class-studies.R' 'class-traits.R' 'class-variants.R' 'data.R' 'ebi_server.R' 'generics.R' 'get_associations.R' 'get_metadata.R' 'get_studies.R' 'get_traits.R' 'get_variants.R' 'gwasrapidd-package.R' 'id_mapping.R' 'list_joins.R' 'missing.R' 'parse-associations.R' 'parse-studies.R' 'parse-traits.R' 'parse-utils.R' 'parse-variants.R' 'post-studies.R' 'post-traits.R' 'post-variants.R' 'recursive_apply.R' 'request.R' 's4-utils.R' 'sure.R' 'tests.R' 'utils-pipe.R' 'utils.R' 'wrappers.R' 'write_xlsx.R' |
| VignetteBuilder: | knitr |
| biocViews: | ThirdPartyClient, BiomedicalInformatics, GenomeWideAssociation, SNP |
| NeedsCompilation: | no |
| Packaged: | 2025-05-31 15:39:47 UTC; rmagno |
| Author: | Ramiro Magno  [aut, cre],
Ana-Teresa Maia
[aut],
CINTESIS [cph, fnd],
Pattern Institute
[aut, cre],
Ana-Teresa Maia
[aut],
CINTESIS [cph, fnd],
Pattern Institute  [cph, fnd] [cph, fnd] |
| Maintainer: | Ramiro Magno <rmagno@pattern.institute> |
| Repository: | CRAN |
| Date/Publication: | 2025-05-31 15:50:02 UTC |
gwasrapidd: 'REST' 'API' Client for the 'NHGRI'-'EBI' 'GWAS' Catalog
Description
![]()
'GWAS' R 'API' Data Download. This package provides easy access to the 'NHGRI'-'EBI' 'GWAS' Catalog data by accessing the 'REST' 'API' https://www.ebi.ac.uk/gwas/rest/docs/api/.
Author(s)
Maintainer: Ramiro Magno rmagno@pattern.institute (ORCID)
Authors:
Ana-Teresa Maia maia.anateresa@gmail.com (ORCID)
Other contributors:
CINTESIS [copyright holder, funder]
Pattern Institute (04jrgd746) [copyright holder, funder]
See Also
Useful links:
Report bugs at https://github.com/ramiromagno/gwasrapidd/issues
Pipe operator
Description
See magrittr::%>% for details.
Value
The same as the rhs.
Examples
c(1,2,3) %>% mean()
Adds an element named <obj_type> to content sub-element.
Description
This function takes a list, sees if it has an element named 'content' and
then checks if content contains an element named obj_type: if it does
not contain an element as passed in obj_type this function adds it,
otherwise it leaves the list obj untouched. The contents of 'content'
are set as value of <obj>$content$<obj_type>.
Usage
add_object_tier(obj, obj_type)
Arguments
obj |
A list. |
obj_type |
A non-empty string. |
Value
A list containing the <obj>$content$<obj_type>, whose value is
either NULL if it had not any value before or the value of
<obj>$content.
Extract allele names from strings of the form rs123-G
Description
This function parses strings of the form "rs123-G" and returns
the name of the allele; it uses the regex ([ATCG]+)$.
Usage
allele_name(risk_allele_names)
Arguments
risk_allele_names |
Value
A character vector of allele names.
Creates an ancestral groups table.
Description
Creates a ancestral groups table.
Usage
ancestral_groups_tbl(
study_id = character(),
ancestry_id = integer(),
ancestral_group = character()
)
Arguments
study_id |
GWAS Catalog study accession identifier. |
ancestry_id |
Ancestry identifier. |
ancestral_group |
Ancestral group. |
Value
A tibble whose columns are the named arguments
to the function.
Creates an ancestries table.
Description
Creates an ancestries table.
Usage
ancestries_tbl(
study_id = character(),
ancestry_id = integer(),
type = character(),
number_of_individuals = integer()
)
Arguments
study_id |
GWAS Catalog study accession identifier. |
ancestry_id |
Ancestry identifier. |
type |
Type of cohort sample, either |
number_of_individuals |
Number of individuals in the cohort sample. |
Value
A tibble whose columns are the named arguments
to the function.
Map an association id to a study id
Description
Map an association accession identifier to a study accession identifier.
Usage
association_to_study(association_id, verbose = FALSE, warnings = TRUE)
Arguments
association_id |
A character vector of association accession identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the association identifier and the second column is the study identifier.
Examples
## Not run:
# Map GWAS association identifiers to study identifiers
association_to_study(c('24300097', '24299759'))
## End(Not run)
Map an association id to an EFO trait id
Description
Map an association accession identifier to an EFO trait id.
Usage
association_to_trait(association_id, verbose = FALSE, warnings = TRUE)
Arguments
association_id |
A character vector of association accession identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the association identifier and the second column is the EFO trait identifier.
Examples
## Not run:
# Map GWAS association identifiers to EFO trait identifiers
association_to_trait(c('24300097', '24299759'))
## End(Not run)
Map an association id to a variant id
Description
Map an association accession identifier to a variant identifier.
Usage
association_to_variant(association_id, verbose = FALSE, warnings = TRUE)
Arguments
association_id |
A character vector of association accession identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the association identifier and the second column is the variant identifier.
Examples
## Not run:
# Map GWAS association identifiers to variant identifiers
association_to_variant(c('24300097', '24299759'))
## End(Not run)
Constructor for the S4 associations object.
Description
Constructor for the S4 associations object.
Usage
associations(
associations = associations_tbl(),
loci = loci_tbl(),
risk_alleles = risk_alleles_tbl(),
genes = reported_genes_tbl(),
ensembl_ids = ensembl_ids_tbl(),
entrez_ids = entrez_ids_tbl()
)
Arguments
associations |
An |
loci |
A |
risk_alleles |
A |
genes |
A |
ensembl_ids |
A |
entrez_ids |
A |
Value
An object of class associations.
An S4 class to represent a set of GWAS Catalog associations
Description
The association object consists of six slots, each a table
(tibble), that combined form a relational database of a
subset of GWAS Catalog associations. Each association is an observation (row)
in the associations table — main table. All tables have the column
association_id as primary key.
Slots
associationsA
tibblelisting associations. Columns:- association_id
GWAS Catalog association accession identifier, e.g.,
"20250".- pvalue
Reported p-value for strongest variant risk or effect allele.
- pvalue_description
Information describing context of p-value.
- pvalue_mantissa
Mantissa of p-value.
- pvalue_exponent
Exponent of p-value.
- multiple_snp_haplotype
Whether the association is for a multi-SNP haplotype.
- snp_interaction
Whether the association is for a SNP-SNP interaction.
- snp_type
Whether the SNP has previously been reported. Either
'known'or'novel'.- risk_frequency
Reported risk/effect allele frequency associated with strongest SNP in controls.
- standard_error
Standard error of the effect size.
- range
Reported 95% confidence interval associated with strongest SNP risk allele, along with unit in the case of beta coefficients. If 95% CIs have not been not reported, these are estimated using the standard error, when available.
- or_per_copy_number
Reported odds ratio (OR) associated with strongest SNP risk allele. Note that all ORs included in the Catalog are >1.
- beta_number
Beta coefficient associated with strongest SNP risk allele.
- beta_unit
Beta coefficient unit.
- beta_direction
Beta coefficient direction, either
'decrease'or'increase'.- beta_description
Additional beta coefficient comment.
- last_mapping_date
Last time this association was mapped to Ensembl.
- last_update_date
Last time this association was updated.
lociA
tibblelisting loci. Columns:- association_id
GWAS Catalog association accession identifier, e.g.,
"20250".- locus_id
A locus identifier referring to a single variant locus or to a multi-loci entity such as a multi-SNP haplotype.
- haplotype_snp_count
Number of variants per locus. Most loci are single-SNP loci, i.e., there is a one to one relationship between a variant and a
locus_id(haplotype_snp_count == NA). There are however cases of associations involving multiple loci at once, such as SNP-SNP interactions and multi-SNP haplotypes. This is signalled in the columns:multiple_snp_haplotypeandsnp_interactionwith valueTRUE.- description
Description of the locus identifier, e.g.,
'Single variant',SNP x SNP interaction, or3-SNP Haplotype.
risk_allelesA
tibblelisting risk alleles. Columns:- association_id
GWAS Catalog association accession identifier, e.g.,
"20250".- locus_id
A locus identifier referring to a single variant locus or to a multi-loci entity such as a multi-SNP haplotype.
- variant_id
Variant identifier, e.g.,
'rs1333048'.- risk_allele
Risk allele or effect allele.
- risk_frequency
Reported risk/effect allele frequency associated with strongest SNP in controls (if not available among all controls, among the control group with the largest sample size). If the associated locus is a haplotype the haplotype frequency will be extracted.
- genome_wide
Whether this variant allele has been part of a genome-wide study or not.
- limited_list
Undocumented.
genesA
tibblelisting author reported genes. Columns:- association_id
GWAS Catalog association accession identifier, e.g.,
"20250".- locus_id
A locus identifier referring to a single variant locus or to a multi-loci entity such as a multi-SNP haplotype.
- gene_name
Gene symbol according to HUGO Gene Nomenclature (HGNC).
ensembl_idsA
tibblelisting Ensembl gene identifiers. Columns:- association_id
GWAS Catalog association accession identifier, e.g.,
"20250".- locus_id
A locus identifier referring to a single variant locus or to a multi-loci entity such as a multi-SNP haplotype.
- gene_name
Gene symbol according to HUGO Gene Nomenclature (HGNC).
- ensembl_id
The Ensembl identifier of an Ensembl gene, see Section Gene annotation in Ensembl for more information.
entrez_idsA
tibblelisting Entrez gene identifiers. Columns:- association_id
GWAS Catalog association accession identifier, e.g.,
"20250".- locus_id
A locus identifier referring to a single variant locus or to a multi-loci entity such as a multi-SNP haplotype.
- gene_name
Gene symbol according to HUGO Gene Nomenclature (HGNC).
- entrez_id
The Entrez identifier of a gene, see ref. doi:10.1093/nar/gkq1237 for more information.
Drop any NA associations.
Description
This function takes an associations S4 object and removes any association
identifiers that might have been NA. This ensures that there is always a
non-NA association_id value in all tables. This is important as the
association_id is the primary key.
Usage
associations_drop_na(s4_associations)
Arguments
s4_associations |
An object of class associations. |
Value
An object of class associations.
Creates an associations table.
Description
Creates an associations table.
Usage
associations_tbl(
association_id = character(),
pvalue = double(),
pvalue_description = character(),
pvalue_mantissa = integer(),
pvalue_exponent = integer(),
multiple_snp_haplotype = logical(),
snp_interaction = logical(),
snp_type = character(),
risk_frequency = double(),
standard_error = double(),
range = character(),
or_per_copy_number = double(),
beta_number = double(),
beta_unit = character(),
beta_direction = character(),
beta_description = character(),
last_mapping_date = lubridate::ymd_hms(),
last_update_date = lubridate::ymd_hms()
)
Arguments
association_id |
A character vector of association identifiers. |
pvalue |
A numeric vector of p-values. |
pvalue_description |
A character vector of p-value context descriptions. |
pvalue_mantissa |
An integer vector of p-value mantissas. |
pvalue_exponent |
An integer vector of p-value exponents. |
multiple_snp_haplotype |
A logical vector. |
snp_interaction |
A logical vector. |
snp_type |
A character vector indicating SNP novelty: 'novel' or 'known'. |
standard_error |
A numeric vector of standard errors. |
range |
A character vector of free text descriptions of confidence intervals. |
or_per_copy_number |
A numeric vector of odds ratios. |
beta_number |
A numeric vector of beta coefficients. |
beta_unit |
A character vector of beta coefficient units. |
beta_direction |
A character vector of beta coefficient directions. |
beta_description |
A character vector of beta descriptions. |
last_mapping_date |
A |
last_update_date |
A |
Value
A tibble whose columns are the named arguments
to the function.
Bind GWAS Catalog objects
Description
Binds together GWAS Catalog objects of the same class. Note that
bind() preserves duplicates whereas
union does not.
Usage
bind(x, ...)
Arguments
x |
An object of class: studies, associations, variants, or traits. |
... |
Objects of the same class as |
Value
An object of the same class as x.
Examples
# Join two studies objects.
bind(studies_ex01, studies_ex02)
# Join two associations objects.
bind(associations_ex01, associations_ex02)
# Join two variants objects.
bind(variants_ex01, variants_ex02)
# Join two traits objects.
bind(traits_ex01, traits_ex02)
Get all child terms of this trait in the EFO hierarchy
Description
Get all child terms of this trait in the EFO hierarchy
Usage
child_efo_ids(
efo_id,
verbose = FALSE,
warnings = TRUE,
page_size = 20L,
progress_bar = TRUE
)
Arguments
efo_id |
A EFO identifier. |
verbose |
A |
warnings |
A |
page_size |
An |
progress_bar |
Whether to show a progress bar as the paginated resources are retrieved. |
Value
A character vector of EFO identifiers.
Does a string contain a question mark?
Description
Find which strings contain a question mark. This function uses the following
regular expression: [\?].
Usage
contains_question_mark(str, convert_NA_to_FALSE = TRUE)
Arguments
str |
A character vector of strings. |
convert_NA_to_FALSE |
Whether to treat |
Value
A logical vector.
Creates a countries table.
Description
Creates a countries table. This
function is used internally to create both the countries_of_origin and
countries_of_recruitment slots of a studies object.
Usage
countries_tbl(
study_id = character(),
ancestry_id = integer(),
country_name = character(),
major_area = character(),
region = character()
)
Arguments
study_id |
GWAS Catalog study accession identifier. |
ancestry_id |
Ancestry identifier. |
country_name |
Country name, according to The United Nations M49 Standard of Geographic Regions. |
major_area |
Region name, according to The United Nations M49 Standard of Geographic Regions. |
region |
Sub-region name, according to The United Nations M49 Standard of Geographic Regions. |
Value
A tibble whose columns are the named arguments
to the function.
Convert a cytogenetic band string to genomic coordinates.
Description
This function uses the provided cytogenetic_bands
dataframe to convert cytogenetic band names to genomic coordinates.
Usage
cytogenetic_band_to_genomic_range(bands)
Arguments
bands |
A |
Value
A dataframe of genomic ranges. Columns are: chromosome, start and end. Each row corresponds to a queried cytogenetic band (in the same order as queried).
GRCh38 human cytogenetic bands.
Description
A dataset containing the GRCh38 human cytogenetic bands and their genomic coordinates.
Usage
cytogenetic_bands
Format
A data frame with 862 rows and 8 variables:
- cytogenetic_band
Cytogenetic band name. See Cytogenetic Nomenclature below.
- chromosome
Chromosome name: 1 through 22 (the autosomes), X or Y.
- start
Genomic start position of the cytogenetic band. Starts at 1.
- end
Genomic end position of the cytogenetic band. End position is included in the band interval.
- length
Length of the genomic interval of cytogenetic band.
- assembly
Assembly version, should be 'GRCh38'.
- stain
Giemsa stain results: Giemsa negative,
'gneg'; Giemsa positive, of increasing intensities,'gpos25','gpos50','gpos75', and'gpos100'; centromeric region,'acen'; heterochromatin, either pericentric or telomeric,'gvar'; and short arm of acrocentric chromosomes 13, 14, 15, 21, and 22 are coded as'stalk'.- last_download_date
Time stamp of last time this dataset was downloaded from Ensembl.
Details
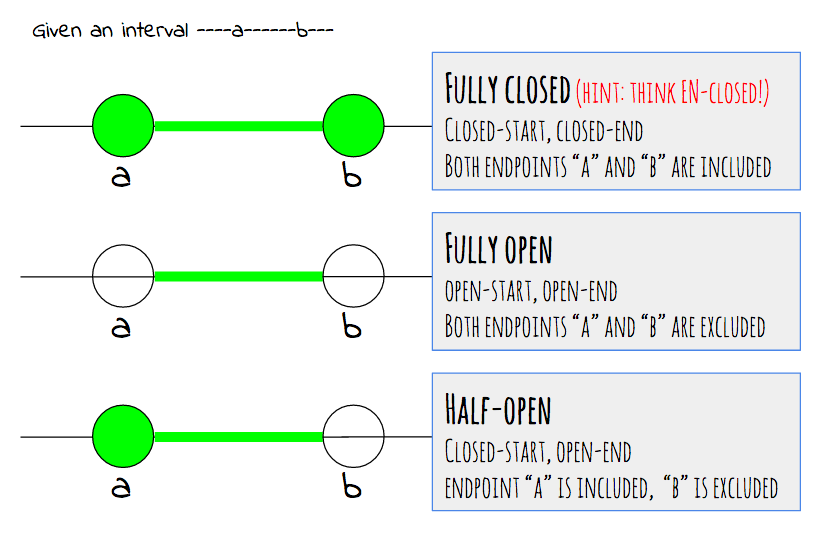
Genomic coordinates are for fully closed intervals.
{kind=link}
Cytogenetic Nomenclature
Cytogenetic bands are numbered from the centromere outwards in both directions towards the telomeres on the shorter p arm and the longer q arm.
The first number or letter represents the chromosome. Chromosomes 1 through 22 (the autosomes) are designated by their chromosome number. The sex chromosomes are designated by X or Y. The next letter represents the arm of the chromosome: p or q.
The numbers cannot be read in the normal decimal numeric system e.g. 36, but rather 3-6 (region 3 band 6). Counting starts at the centromere as region 1 (or 1-0), to 11 (1-1) to 21 (2-1) to 22 (2-2) etc. Subbands are added in a similar way, e.g. 21.1 to 21.2, if the bands are small or only appear at a higher resolution.
Source
https://rest.ensembl.org/info/assembly/homo_sapiens?content-type=application/json&bands=1
Converts an empty vector to a scalar NA
Description
This function converts an empty vector to a scalar NA of the same type as the input.
Usage
empty_to_na(x)
Arguments
x |
An atomic vector, of one of these types: |
Value
An atomic vector of the same type as x. If x is empty
then NA is returned, otherwise x is returned as is.
Creates an Ensembl gene identifiers' table.
Description
Creates an Ensembl gene identifiers' table.
Usage
ensembl_ids_tbl(
association_id = character(),
locus_id = integer(),
gene_name = character(),
ensembl_id = character()
)
Arguments
association_id |
A character vector of association identifiers. |
locus_id |
An integer vector of locus identifiers. |
gene_name |
A character vector of gene symbol according to HUGO Gene Nomenclature (HGNC). |
ensembl_id |
A character vector of Ensembl identifiers. |
Value
A tibble whose columns are the named arguments
to the function.
Creates an Entrez gene identifiers' table.
Description
Creates an Entrez gene identifiers' table.
Usage
entrez_ids_tbl(
association_id = character(),
locus_id = integer(),
gene_name = character(),
entrez_id = character()
)
Arguments
association_id |
A character vector of association identifiers. |
locus_id |
An integer vector of locus identifiers. |
gene_name |
A character vector of gene symbol according to HUGO Gene Nomenclature (HGNC). |
entrez_id |
A character vector of Entrez identifiers. |
Value
A tibble whose columns are the named arguments
to the function.
Grows vectors to match longest vector
Description
This function determines the longest vector and pads the shorter ones by adding NAs until they match the longest vector.
Usage
equal_length(lst_of_vectors)
Arguments
lst_of_vectors |
A list of atomic vectors. |
Value
A list of atomic vectors of the same length.
Check if a variant exists in the Catalog.
Description
This function attempts to get a variant by its variant identifier and checks the response code. If the response code is 200 then the response has been successful, meaning that the variant does exist in the GWAS Catalog. If the response is 404 then the variant is not found in the Catalog database. Other errors are mapped to NA.
Usage
exists_variant(variant_id = NULL, verbose = FALSE, page_size = 20L)
Arguments
variant_id |
A character vector of GWAS Catalog variant identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A named logical vector, TRUE indicates that the variant does
exist in the Catalog, FALSE otherwise. NA codes other types
of errors. The names of the vector are the variant identifiers passed as
variant_id.
Examples
exists_variant('rs12345')
exists_variant('rs11235813')
Extract association identifiers from URLs
Description
This function extracts association identifiers from URLs of the form:
".*{association_id}$".
Usage
extract_association_id(urls)
Arguments
urls |
A character vector of URLs of the form
|
Value
A character vector of association identifiers.
Filter GWAS Catalog objects by identifier.
Description
Use filter_by_id to filter GWAS Catalog objects by their respective
identifier (id).
Usage
filter_by_id(x, id)
Arguments
x |
An object of class either studies, associations, variants, or traits. |
id |
Identifier. |
Value
Returns an object of class either studies, associations, variants, or traits.
Filter variants by standard human chromosomes.
Description
This function filters a variants object by standard human chromosomes, i.e.,
1–22, X and Y. In addition to these chromosomes, some variants retrieved
from the GWAS Catalog might be also mapped to non-standard locations, such as
GRC assembly patches, haplotype (HAPs) or pseudo autosomal regions (PARs).
When this happens the main table variants includes rows for these
cases too. This function removes these.
Usage
filter_variants_by_standard_chromosomes(
s4_variants,
chromosomes = c(seq_len(22), "X", "Y", "MT")
)
Arguments
s4_variants |
An object of class variants. |
chromosomes |
A character vector of valid chromosome names. Default is autosomal chromosomes 1 thru 22 and, X, Y, and MT. |
Value
An object of class variants.
gwasrapidd entities' examples
Description
These are examples of GWAS Catalog entities shipped with gwasrapidd:
Usage
studies_ex01
studies_ex02
associations_ex01
associations_ex02
variants_ex01
variants_ex02
traits_ex01
traits_ex02
Format
- studies_ex01
An S4 studies object of 2 studies:
'GCST001585'and'GCST003985'.- studies_ex02
An S4 studies object of 2 studies:
'GCST001585'and'GCST006655'.- associations_ex01
An S4 associations object of 4 associations:
'22509','22505','19537565'and'19537593'.- associations_ex02
An S4 associations object of 3 associations:
'19537593','31665940'and'34944736'.- variants_ex01
An S4 variants object of 3 variants:
'rs146992477','rs56261590'and'rs4725504'.- variants_ex02
An S4 variants object of 4 variants:
'rs56261590','rs4725504','rs11099757'and'rs16871509'.- traits_ex01
An S4 traits object of 3 traits:
'EFO_0004884','EFO_0004343'and'EFO_0005299'.- traits_ex02
An S4 traits object of 4 traits:
'EFO_0007845','EFO_0004699','EFO_0004884'and'EFO_0004875'.
An object of class studies of length 1.
An object of class associations of length 1.
An object of class associations of length 1.
An object of class variants of length 1.
An object of class variants of length 1.
An object of class traits of length 1.
An object of class traits of length 1.
Get a GWAS Catalog resource
Description
This function gets a GWAS Catalog by URL endpoint. The response must correspond to one of the four types of entities: studies, associations, variants or traits.
Usage
gc_get(
resource_url,
base_url = gwas_rest_api_base_url,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
resource_url |
Endpoint URL. The endpoint is internally appended to the
|
base_url |
The GWAS REST API base URL (one should not need to change its default value). |
verbose |
Whether to be chatty. |
warnings |
Whether to print warnings. |
page_size |
Page parameter used in the URL endpoint. |
Value
A normalised JSON-list corresponding to either studies, associations, variants or traits.
Request a GWAS Catalog REST API endpoint
Description
Performs a GET request on the specified resource_url.
Usage
gc_request(
resource_url = "/",
base_url = gwas_rest_api_base_url,
verbose = FALSE,
warnings = TRUE,
flatten = FALSE
)
Arguments
resource_url |
Endpoint URL. The endpoint is internally appended to the
|
base_url |
The GWAS REST API base URL (one should not need to change its default value). |
verbose |
Whether to be chatty. |
warnings |
Whether to print warnings. |
flatten |
Whether to flatten out the list returned by
|
Value
A list four named elements:
- url
The URL endpoint.
- response_code
- status
A string describing the status of the response obtained. It is "OK" if everything went OK or some other string describing the problem otherwise.
- content
The parsed JSON as a nested list, as returned by
jsonlite::fromJSON.
Request a paginated GWAS Catalog REST API endpoint
Description
Performs a GET request on the specified resource_url and all its
pages.
Usage
gc_request_all(
resource_url = "/",
base_url = gwas_rest_api_base_url,
page_size = 20L,
verbose = FALSE,
warnings = TRUE,
progress_bar = TRUE
)
Arguments
resource_url |
Endpoint URL. The endpoint is internally appended to the
|
base_url |
The GWAS REST API base URL (one should not need to change its default value). |
page_size |
Page parameter used in the URL endpoint. |
verbose |
Whether to be chatty. |
warnings |
Whether to print warnings. |
progress_bar |
Whether to show a progress bar as the paginated resources are retrieved. |
Value
A list four named elements:
- url
The URL endpoint.
- response_code
- status
A string describing the status of the response obtained. It is "OK" if everything went OK or some other string describing the problem otherwise.
- content
The parsed JSON as a nested list, as returned by
jsonlite::fromJSON.
Creates a genomic contexts table.
Description
Creates a genomic contexts table.
Usage
genomic_contexts_tbl(
variant_id = character(),
gene_name = character(),
chromosome_name = character(),
chromosome_position = integer(),
distance = integer(),
is_mapped_gene = logical(),
is_closest_gene = logical(),
is_intergenic = logical(),
is_upstream = logical(),
is_downstream = logical(),
source = character(),
mapping_method = character()
)
Arguments
variant_id |
A character vector of variant identifiers. |
gene_name |
A character vector of gene symbols according to HUGO Gene Nomenclature (HGNC). |
chromosome_name |
A character vector of chromosome names. |
chromosome_position |
An integer vector of chromosome positions. |
distance |
An integer vector of genomic positions. |
is_closest_gene |
A logical vector. |
is_intergenic |
A logical vector. |
is_upstream |
A logical vector. |
is_downstream |
A logical vector. |
source |
A character vector of gene mapping sources. |
mapping_method |
A character vector of gene mapping methods. |
Value
A tibble whose columns are the named arguments
to the function.
Creates a genotyping technologies table.
Description
Creates a genotyping technologies table.
Usage
genotyping_techs_tbl(
study_id = character(),
genotyping_technology = character()
)
Arguments
study_id |
GWAS Catalog study accession identifier. |
genotyping_technology |
Genotyping technology. |
Value
A tibble whose columns are the named arguments
to the function.
Get GWAS Catalog associations
Description
Retrieves associations via the NHGRI-EBI GWAS Catalog REST API. The REST
API is queried multiple times with the criteria passed as arguments (see
below). By default all associations that match the criteria supplied in the
arguments are retrieved: this corresponds to the default option
set_operation set to 'union'. If you rather have only the
associations that match simultaneously all criteria provided, then set
set_operation to 'intersection'.
Usage
get_associations(
study_id = NULL,
association_id = NULL,
variant_id = NULL,
efo_id = NULL,
pubmed_id = NULL,
efo_trait = NULL,
set_operation = "union",
interactive = TRUE,
verbose = FALSE,
warnings = TRUE
)
Arguments
study_id |
A |
association_id |
A |
variant_id |
A |
efo_id |
A character vector of EFO identifiers. |
pubmed_id |
An |
efo_trait |
A |
set_operation |
Either |
interactive |
A logical. If all associations are requested, whether to ask interactively if we really want to proceed. |
verbose |
A |
warnings |
A |
Details
Please note that all search criteria are vectorised, thus allowing for batch
mode search, e.g., one can search by multiple variant identifiers at once by
passing a vector of identifiers to variant_id.
Value
An associations object.
Examples
## Not run:
# Get an association by study identifier
get_associations(study_id = 'GCST001085', warnings = FALSE)
# Get an association by association identifier
get_associations(association_id = '25389945', warnings = FALSE)
# Get associations by variant identifier
get_associations(variant_id = 'rs3798440', warnings = FALSE)
# Get associations by EFO trait identifier
get_associations(efo_id = 'EFO_0005537', warnings = FALSE)
## End(Not run)
Get all GWAS Catalog associations
Description
Gets all associations. Beware this can take a few hours!
Usage
get_associations_all(verbose = FALSE, warnings = TRUE, page_size = 20L)
Arguments
verbose |
A |
warnings |
A |
page_size |
An |
Value
A associations object.
Get GWAS Catalog associations by their association identifiers
Description
Gets associations by GWAS Catalog internal association identifiers.
Usage
get_associations_by_association_id(
association_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
association_id |
A |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A associations object.
Get GWAS Catalog associations by EFO identifier
Description
Gets associations whose phenotypic trait is matched by EFO identifiers.
Usage
get_associations_by_efo_id(
efo_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
efo_id |
A character vector of EFO identifiers. |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A associations object.
Get GWAS Catalog associations by EFO traits
Description
Gets associations that match EFO trait description.
Usage
get_associations_by_efo_trait(
efo_trait = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
efo_trait |
A |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A associations object.
Get GWAS Catalog associations by PubMed identifiers
Description
Gets associations whose associated publications match PubMed identifiers.
Usage
get_associations_by_pubmed_id(
pubmed_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
pubmed_id |
An |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A associations object.
Get GWAS Catalog associations by study identifiers
Description
Gets associations by GWAS Catalog internal study identifiers.
Usage
get_associations_by_study_id(
study_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
study_id |
A |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A associations object.
Get GWAS Catalog associations by variant identifiers
Description
Gets associations by variant identifiers.
Usage
get_associations_by_variant_id(
variant_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
variant_id |
A |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A associations object.
Get all child terms of this trait in the EFO hierarchy
Description
Get all child terms of this trait in the EFO hierarchy
Usage
get_child_efo(
efo_id,
verbose = FALSE,
warnings = TRUE,
page_size = 20L,
progress_bar = TRUE
)
Arguments
efo_id |
A character vector of EFO identifiers. |
verbose |
A |
warnings |
A |
page_size |
An |
progress_bar |
Whether to show a progress bar as the paginated resources are retrieved. |
Value
A named list whose values are character vectors of EFO identifiers.
Examples
## Not run:
get_child_efo(c('EFO_0004884', 'EFO_0004343', 'EFO_0005299'))
## End(Not run)
Get GWAS Catalog metadata
Description
Provides a list of the resources the GWAS Catalog data is currently mapped against: Ensembl release number, Genome build version and dbSNP version.In addition, the date since this combination of resource versions has been in use is also returned.
Usage
get_metadata(verbose = FALSE, warnings = TRUE)
Arguments
verbose |
Whether to be chatty. |
warnings |
Whether to trigger a warning if the request is not successful. |
Value
-
ensembl_release_number: Ensembl release number; -
genome_build_version: Genome build version; -
dbsnp_version: dbSNP version. -
usage_start_date: Date since this combination of resource versions has been in use.
Examples
## Not run:
get_metadata(warnings = FALSE)
## End(Not run)
Get GWAS Catalog studies
Description
Retrieves studies via the NHGRI-EBI GWAS Catalog REST API. The REST
API is queried multiple times with the criteria passed as arguments (see
below). By default all studies that match the criteria supplied in the
arguments are retrieved: this corresponds to the default option
set_operation set to 'union'. If you rather have only the
studies that match simultaneously all criteria provided, then set
set_operation to 'intersection'.
Usage
get_studies(
study_id = NULL,
association_id = NULL,
variant_id = NULL,
efo_id = NULL,
pubmed_id = NULL,
user_requested = NULL,
full_pvalue_set = NULL,
efo_uri = NULL,
efo_trait = NULL,
reported_trait = NULL,
set_operation = "union",
interactive = TRUE,
verbose = FALSE,
warnings = TRUE
)
Arguments
study_id |
A character vector of GWAS Catalog study accession identifiers. |
association_id |
A character vector of GWAS Catalog association identifiers. |
variant_id |
A character vector of GWAS Catalog variant identifiers. |
efo_id |
A character vector of EFO identifiers. |
pubmed_id |
An integer vector of PubMed identifiers. |
user_requested |
A |
full_pvalue_set |
A |
efo_uri |
A character vector of EFO URIs. |
efo_trait |
A character vector of
EFO trait descriptions, e.g.,
|
reported_trait |
A character vector of phenotypic traits as reported by the original authors of the study. |
set_operation |
Either |
interactive |
A logical. If all studies are requested, whether to ask interactively if we really want to proceed. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Details
Please note that all search criteria are vectorised, thus allowing for batch
mode search, e.g., one can search by multiple variant identifiers at once by
passing a vector of identifiers to variant_id.
Value
A studies object.
Examples
## Not run:
# Get a study by its accession identifier
get_studies(study_id = 'GCST001085', warnings = FALSE)
# Get a study by association identifier
get_studies(association_id = '25389945', warnings = FALSE)
# Get studies by variant identifier
get_studies(variant_id = 'rs3798440', warnings = FALSE)
# Get studies by EFO trait identifier
get_studies(efo_id = 'EFO_0005537', warnings = FALSE)
## End(Not run)
Get all GWAS Catalog studies
Description
Gets all studies. Beware this can take several minutes!
Usage
get_studies_all(verbose = FALSE, warnings = TRUE, page_size = 20L)
Arguments
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A studies object.
Get GWAS Catalog studies by association identifiers
Description
Gets studies by GWAS Catalog internal association identifiers.
Usage
get_studies_by_association_id(
association_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
association_id |
A character vector of GWAS Catalog association identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An |
Value
A studies object.
Get GWAS Catalog studies by EFO identifier
Description
Gets studies whose phenotypic trait is matched by EFO identifiers.
Usage
get_studies_by_efo_id(
efo_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
efo_id |
A character vector of EFO identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A studies object.
Get GWAS Catalog studies by EFO traits
Description
Gets studies that match EFO trait description.
Usage
get_studies_by_efo_trait(
efo_trait = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
efo_trait |
A |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A studies object.
Get GWAS Catalog studies by EFO URIs
Description
Gets studies that match EFO URI.
Usage
get_studies_by_efo_uri(
efo_uri = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
efo_uri |
A character vector of EFO URIs. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A studies object.
Get GWAS Catalog studies by full summary statistics criterion
Description
Gets studies that either have full summary statistics or studies that do not have it.
Usage
get_studies_by_full_pvalue_set(
full_pvalue_set = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
full_pvalue_set |
A |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A studies object.
Get GWAS Catalog studies by PubMed identifiers
Description
Gets studies whose associated publications match PubMed identifiers.
Usage
get_studies_by_pubmed_id(
pubmed_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
pubmed_id |
An integer vector of PubMed identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A studies object.
Get GWAS Catalog studies by reported traits
Description
Gets studies that match the reported traits, as reported by the original authors' of the study.
Usage
get_studies_by_reported_trait(
reported_trait = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
reported_trait |
A character vector of phenotypic traits as reported by the original authors' the study. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A studies object.
Get GWAS Catalog studies by study identifiers
Description
Gets studies by GWAS Catalog internal study identifiers.
Usage
get_studies_by_study_id(
study_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
study_id |
A character vector of GWAS Catalog study accession identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A studies object.
Get GWAS Catalog studies that have been requested by users or not
Description
Gets studies that have either been requested by users of the Catalog or studies that have not been explicitly requested by users.
Usage
get_studies_by_user_requested(
user_requested = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
user_requested |
A |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A studies object.
Get GWAS Catalog studies by variant identifiers
Description
Gets studies by variant identifiers.
Usage
get_studies_by_variant_id(
variant_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
variant_id |
A character vector of GWAS Catalog variant identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A studies object.
Get GWAS Catalog EFO traits
Description
Retrieves traits via the NHGRI-EBI GWAS Catalog REST API. The REST
API is queried multiple times with the criteria passed as arguments (see
below). By default all traits that match the criteria supplied in the
arguments are retrieved: this corresponds to the default option
set_operation set to 'union'. If you rather have only the
traits that match simultaneously all criteria provided, then set
set_operation to 'intersection'.
Usage
get_traits(
study_id = NULL,
association_id = NULL,
efo_id = NULL,
pubmed_id = NULL,
efo_uri = NULL,
efo_trait = NULL,
set_operation = "union",
verbose = FALSE,
warnings = TRUE
)
Arguments
study_id |
A |
association_id |
A |
efo_id |
A character vector of EFO identifiers. |
pubmed_id |
An |
efo_uri |
A |
efo_trait |
A |
set_operation |
Either |
verbose |
A |
warnings |
A |
Details
Please note that all search criteria are vectorised, thus allowing for batch
mode search, e.g., one can search by multiple trait identifiers at once by
passing a vector of identifiers to efo_id.
Value
A traits object.
Examples
## Not run:
# Get traits by study identifier
get_traits(study_id = 'GCST001085', warnings = FALSE)
# Get traits by association identifier
get_traits(association_id = '25389945', warnings = FALSE)
# Get a trait by its EFO identifier
get_traits(efo_id = 'EFO_0005537', warnings = FALSE)
## End(Not run)
Get all GWAS Catalog EFO traits
Description
Gets all EFO traits. Beware this can take several minutes!
Usage
get_traits_all(verbose = FALSE, warnings = TRUE, page_size = 20L)
Arguments
verbose |
A |
warnings |
A |
page_size |
An |
Value
A traits object.
Get GWAS Catalog traits by association identifiers
Description
Gets traits by GWAS Catalog internal association identifiers.
Usage
get_traits_by_association_id(
association_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
association_id |
A |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A traits object.
Get GWAS Catalog traits by EFO identifier
Description
Gets traits whose phenotypic trait is matched by EFO identifiers.
Usage
get_traits_by_efo_id(
efo_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
efo_id |
A character vector of EFO identifiers. |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A traits object.
Get GWAS Catalog traits by EFO traits
Description
Gets traits that match EFO trait description.
Usage
get_traits_by_efo_trait(
efo_trait = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
efo_trait |
A |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A traits object.
Get GWAS Catalog traits by EFO URIs
Description
Gets traits that match EFO URI.
Usage
get_traits_by_efo_uri(
efo_uri = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
efo_uri |
A |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A traits object.
Get GWAS Catalog traits by PubMed identifiers
Description
Gets traits whose associated publications match PubMed identifiers.
Usage
get_traits_by_pubmed_id(
pubmed_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
pubmed_id |
An |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A traits object.
Get GWAS Catalog traits by study identifiers
Description
Gets traits by GWAS Catalog internal study identifiers.
Usage
get_traits_by_study_id(
study_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
study_id |
A |
verbose |
A |
warnings |
A |
page_size |
An |
Value
A traits object.
Get GWAS Catalog variants
Description
Retrieves variants via the NHGRI-EBI GWAS Catalog REST API. The REST
API is queried multiple times with the criteria passed as arguments (see
below). By default all variants that match the criteria supplied in the
arguments are retrieved: this corresponds to the default option
set_operation set to 'union'. If you rather have only the
variants that match simultaneously all criteria provided, then set
set_operation to 'intersection'.
Usage
get_variants(
study_id = NULL,
association_id = NULL,
variant_id = NULL,
efo_id = NULL,
pubmed_id = NULL,
genomic_range = NULL,
cytogenetic_band = NULL,
gene_name = NULL,
efo_trait = NULL,
reported_trait = NULL,
set_operation = "union",
interactive = TRUE,
std_chromosomes_only = TRUE,
verbose = FALSE,
warnings = TRUE
)
Arguments
study_id |
A character vector of GWAS Catalog study accession identifiers. |
association_id |
A character vector of GWAS Catalog association identifiers. |
variant_id |
A character vector of GWAS Catalog variant identifiers. |
efo_id |
A character vector of EFO identifiers. |
pubmed_id |
An integer vector of PubMed identifiers. |
genomic_range |
A named list of three vectors:
The three vectors need to be of the same length so that |
cytogenetic_band |
A character vector of cytogenetic bands of the form
|
gene_name |
Gene symbol according to HUGO Gene Nomenclature (HGNC). |
efo_trait |
A character vector of
EFO trait descriptions, e.g.,
|
reported_trait |
A character vector of phenotypic traits as reported by the original authors of the study. |
set_operation |
Either |
interactive |
A logical. If all variants are requested, whether to ask interactively if we really want to proceed. |
std_chromosomes_only |
Whether to return only variants mapped to standard chromosomes: 1 thru 22, X, Y, and MT. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Details
Please note that all search criteria are vectorised, thus allowing for batch
mode search, e.g., one can search by multiple variant identifiers at once by
passing a vector of identifiers to variant_id.
Value
A variants object.
Examples
# Get variants by study identifier
get_variants(study_id = 'GCST001085', warnings = FALSE)
# Get a variant by its identifier
## Not run:
get_variants(variant_id = 'rs3798440', warnings = FALSE)
## End(Not run)
Get all GWAS Catalog variants
Description
Gets all variants. Beware this can take several minutes!
Usage
get_variants_all(verbose = FALSE, warnings = TRUE, page_size = 20L)
Arguments
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A variants object.
Get GWAS Catalog variants by their association identifiers
Description
Gets variants by GWAS Catalog internal association identifiers.
Usage
get_variants_by_association_id(
association_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
association_id |
A character vector of GWAS Catalog association identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A variants object.
Get GWAS Catalog variants by cytogenetic band.
Description
Gets variants that are mapped onto specific regions as specified by
cytogenetic bands. See the dataframe
cytogenetic_bands for more information on possible
values.
Usage
get_variants_by_cytogenetic_band(
cytogenetic_band = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
cytogenetic_band |
A |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A variants object.
Get GWAS Catalog studies by EFO identifier
Description
Gets variants whose phenotypic trait is matched by EFO identifiers.
Usage
get_variants_by_efo_id(
efo_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
efo_id |
A character vector of EFO identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A variants object.
Get GWAS Catalog variants by EFO traits
Description
Gets variants that match EFO trait description.
Usage
get_variants_by_efo_trait(
efo_trait = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
efo_trait |
A character vector of
EFO trait descriptions, e.g.,
|
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An |
Value
A variants object.
Get GWAS Catalog variants by gene name.
Description
Gets variants whose genomic context includes a specific gene or genes.
Usage
get_variants_by_gene_name(
gene_name = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
gene_name |
A |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A variants object.
Get GWAS Catalog variants by genomic range
Description
Gets variants by genomic range.
Usage
get_variants_by_genomic_range(
chromosome = NULL,
start = NULL,
end = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
chromosome |
A character vector of human chromosome names: autosomal and sexual chromosomes only, i.e., 1–22, X and Y. |
start |
Start position of range (starts at 1). |
end |
End position of range (inclusive). |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A variants object.
Get GWAS Catalog variants by PubMed identifiers
Description
Gets variants whose associated publications match PubMed identifiers.
Usage
get_variants_by_pubmed_id(
pubmed_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
pubmed_id |
An integer vector of PubMed identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A variants object.
Get GWAS Catalog variants by reported traits
Description
Gets variants that match the reported traits, as reported by the original authors' of the study.
Usage
get_variants_by_reported_trait(
reported_trait = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
reported_trait |
A character vector of phenotypic traits as reported by the original authors' the study. Note: this parameter is case sensitive. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A variants object.
Get GWAS Catalog variants by study identifiers
Description
Gets variants by GWAS Catalog internal study identifiers.
Usage
get_variants_by_study_id(
study_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
study_id |
A character vector of GWAS Catalog study accession identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
page_size |
An integer scalar indicating the
page
value to be used in the JSON requests, can be between |
Value
A variants object.
Get GWAS Catalog variants by variant identifiers
Description
Gets variants by variant identifiers.
Usage
get_variants_by_variant_id(
variant_id = NULL,
verbose = FALSE,
warnings = TRUE,
page_size = 20L
)
Arguments
variant_id |
A character vector of GWAS Catalog variant identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings, if any. |
page_size |
An |
Value
A variants object.
Is a string a GWAS Catalog association accession ID?
Description
Find which strings are valid GWAS Catalog association IDs (returns
TRUE). Association IDs are tested against the following regular
expression: ^\\d+$.
Usage
is_association_id(str, convert_NA_to_FALSE = TRUE)
Arguments
str |
A character vector of strings. |
convert_NA_to_FALSE |
Whether to treat |
Value
A logical vector.
Is the GWAS Catalog REST API server reachable?
Description
Check if the EBI server where the GWAS Catalog REST API server is running is
reachable. This function attempts to connect to
https://www.ebi.ac.uk, returning TRUE on
success, and FALSE otherwise. Set chatty = TRUE for a step by
step description of the connection attempt.
Usage
is_ebi_reachable(url = "https://www.ebi.ac.uk", port = 443L, chatty = FALSE)
Arguments
url |
NHGRI-EBI GWAS Catalog server URL. Default is https://www.ebi.ac.uk. You should not need to change this parameter. |
port |
Network port on which to ping the server. You should not need to change this parameter. |
chatty |
Whether to be verbose ( |
Value
A logical value: TRUE if EBI server is reachable, FALSE
otherwise.
Examples
# Check if the GWAS Catalog Server is reachable
is_ebi_reachable() # Returns TRUE or FALSE.
# Check if the GWAS Catalog Server is reachable
# and show exactly at what step is it failing (if that is the case)
is_ebi_reachable(chatty = TRUE)
Is a string an EFO trait ID?
Description
Find which strings are valid EFO trait IDs (returns
TRUE). EFO trait IDs are tested against the following regular
expression: ^EFO_\\d{7}$.
Usage
is_efo_id(str, convert_NA_to_FALSE = TRUE)
Arguments
str |
A character vector of strings. |
convert_NA_to_FALSE |
Whether to treat |
Value
A logical vector.
Is a string an EFO trait ID in the broad sense?
Description
This function is more permissible than is_efo_id.
This function matches EFO trait IDs against the following regular expression:
^\\w+$. This is very forgiving on the input, any sequence of word
characters are ok. This is useful to match EFO identifiers that do not follow
the regex ^EFO_\\d{7}$, such as: 'GO_0097334',
'HP_0001268', 'Orphanet_182098', and 'NCIT_C74532'.
Usage
is_efo_id2(str, convert_NA_to_FALSE = TRUE)
Arguments
str |
A character vector of strings. |
convert_NA_to_FALSE |
Whether to treat |
Value
A logical vector.
Is the GWAS response wrapped in an '_embedded' object?
Description
Checks if the response is wrapped in an '_embedded' object by checking if an element named '_embedded' exists.
Usage
is_embedded(obj)
Arguments
obj |
The response object as return by |
Value
A logical value.
Is string empty or an all whitespace string?
Description
Matches the string vector against "^\\s*$".
Usage
is_empty_str(str, convert_NA_to_FALSE = TRUE)
Arguments
str |
A |
convert_NA_to_FALSE |
Whether to treat |
Value
A logical.
Is a string a human chromosome name?
Description
Find which strings are valid human chromosome names. The valid chromosome
names can be specified via the argument chromosomes.
Usage
is_human_chromosome(
string,
chromosomes = c(seq_len(22), "X", "Y", "MT"),
convert_NA_to_FALSE = TRUE
)
Arguments
string |
A character vector of strings. |
chromosomes |
A character vector of valid chromosome names. Default is autosomal chromosomes 1 thru 22 and, X, Y, and MT. |
convert_NA_to_FALSE |
Whether to treat |
Value
A logical vector.
Is the GWAS response paginated?
Description
Checks if the response is paginated by checking if an element named 'page' exists.
Usage
is_paginated(content)
Arguments
content |
The response content as return by |
Value
A logical value.
Is a string a PubMed ID?
Description
Find which strings are valid PubMed IDs (returns TRUE). PubMed IDs are
tested against the following regular expression: ^\\d+$.
Usage
is_pubmed_id(str, convert_NA_to_FALSE = TRUE)
Arguments
str |
A character vector of strings. |
convert_NA_to_FALSE |
Whether to treat |
Value
A logical vector.
Is a string a valid rsID?
Description
Find which strings are valid SNP reference IDs, i.e., of the form rs[0-9]+. Please note that this only does a syntax validation on the strings. It does not check whether the actual IDs exist in dbSNP.
Usage
is_rs_id(str, convert_NA_to_FALSE = TRUE)
Arguments
str |
A character vector of putative SNP reference IDs of the form rs[0-9]+. |
convert_NA_to_FALSE |
Whether to preserve |
Value
Returns a logical vector of the same length as str,
TRUE for strings that are valid rs IDs, and FALSE otherwise.
Is a string a GWAS Catalog study accession ID?
Description
Find which strings are valid GWAS Catalog study accession IDs (returns
TRUE). Study accession IDs are tested against the following regular
expression: ^GCST\\d+$.
Usage
is_study_id(str, convert_NA_to_FALSE = TRUE)
Arguments
str |
A character vector of strings. |
convert_NA_to_FALSE |
Whether to treat |
Value
A logical vector.
Convert a named list to an S4 object
Description
Convert a named list to an S4 object
Usage
list_to_s4(list, class)
Arguments
list |
list |
class |
character vector indicating the S4 class |
Value
S4 object of class class.
Creates a loci table.
Description
Creates a loci table.
Usage
loci_tbl(
association_id = character(),
locus_id = integer(),
haplotype_snp_count = integer(),
description = character()
)
Arguments
association_id |
A character vector of association identifiers. |
locus_id |
An integer vector of locus identifiers. |
haplotype_snp_count |
An integer vector indicating the number of variants in the haplotype. |
description |
A character vector of descriptions, one per locus identifier. |
Value
A tibble whose columns are the named arguments
to the function.
Joins for lists.
Description
These functions join lists by matching elements by name. In the case of
lst_left_join lst_x is used as reference for traversal and its
names are looked for in lst_y for joining. In the case of
lst_left_join, lst_y is used as reference.
lst_inner_join only uses names common to both lst_x and
lst_y for combining elements.
Usage
lst_left_join(lst_x, lst_y)
lst_right_join(lst_x, lst_y)
lst_inner_join(lst_x, lst_y)
plst_left_join(list_of_lsts)
plst_right_join(list_of_lsts)
plst_inner_join(list_of_lsts)
Arguments
lst_x, lst_y |
lists. |
list_of_lsts |
A list of lists to be joined together. |
Details
The functions plst_left_join, plst_right_join and
plst_inner_join are parallel versions that allow joining more than two
lists easily, i.e., just pass a list of lists to be joined.
Value
A list.
Generate a list to hold GWAS metadata
Description
Creates a list to hold GWAS Catalog metadata.
Usage
metadata_lst(
ensembl_release_number = NA_integer_,
genome_build_version = NA_character_,
dbsnp_version = NA_integer_,
usage_start_date = lubridate::ymd_hms()
)
Arguments
ensembl_release_number |
|
genome_build_version |
|
dbsnp_version |
|
usage_start_date |
Date since this combination of resource versions has been in use. |
Value
A list of 4 named elements as passed as arguments:
ensembl_release_number, genome_build_version,
dbsnp_version and usage_start_date.
Recursively map all missing values to NA.
Description
Traverse a recursive list and map missing values to NA.
Missing values are:
-
NULL -
"NR"
Usage
missing_to_na(lst, na = NA_character_)
Arguments
lst |
A list. |
Details
Note: This function is case sensitive on "NR", i.e., it will leave
"nr" untouched.
Value
A list whose missing values have been replaced by NA.
Number of GWAS Catalog entities
Description
This function returns the number of unique entities in a GWAS Catalog object.
Usage
n(x, unique = FALSE)
## S4 method for signature 'studies'
n(x, unique = FALSE)
## S4 method for signature 'associations'
n(x, unique = FALSE)
## S4 method for signature 'variants'
n(x, unique = FALSE)
## S4 method for signature 'traits'
n(x, unique = FALSE)
Arguments
x |
A studies, an associations, a variants, or a traits object. |
unique |
Whether to count only unique entries ( |
Value
An integer scalar.
Examples
# Determine number of studies
n(studies_ex01)
# Determine number of associations
n(associations_ex01)
# Determine number of variants
n(variants_ex01)
# Determine number of traits
n(traits_ex01)
Normalise a JSON-list.
Description
This function normalises a JSON-list. The concept of JSON-list is here
defined as an ordinary R list object whose elements are either all named or
none is named. These lists map naturally to JSON elements: objects and
arrays. What this function does is wrap certain elements of the nested
list obj in list() to make the tree structure of the obj
list homologous across different responses. This makes all responses of the
same entity type (studies, associations, variants or traits) homologous and
hence joinable with family functions lst_*_join.
Usage
normalise_obj(obj, resource_url)
Arguments
obj |
A JSON-list. This is just an ordinary list as returned by
|
resource_url |
The URL endpoint used to obtain the JSON-list |
Details
This normalisation is GWAS object type specific. The parameter
resource_url should be mappable to either studies, associations,
variants or traits by object_type_from_url.
Value
A normalised JSON-list.
Identify the GWAS object entity from the URL endpoint
Description
This function takes URL endpoints and returns one of: studies, associations, variants or traits.
Usage
object_type_from_url(resource_url)
Arguments
resource_url |
A character vector of GWAS URL endpoints. |
Value
A character vector of either "studies", "associations",
"variants" or "traits".
Browse dbSNP from SNP identifiers.
Description
This function launches the web browser at dbSNP and opens a tab for each SNP identifier.
Usage
open_in_dbsnp(variant_id)
Arguments
variant_id |
A variant identifier, a character vector. |
Value
Returns TRUE if successful. Note however that this
function is run for its side effect.
Examples
open_in_dbsnp('rs56261590')
Browse GTEx from SNP identifiers.
Description
This function launches the web browser at the GTEx Portal and opens a tab for each SNP identifier.
Usage
open_in_gtex(variant_id)
Arguments
variant_id |
A variant identifier, a character vector. |
Value
Returns TRUE if successful. Note however that this
function is run for its side effect.
Examples
open_in_gtex('rs56261590')
Browse GWAS Catalog entities from the GWAS Web Graphical User Interface
Description
This function launches the web browser and opens a tab for each identifier on the GWAS web graphical user interface: https://www.ebi.ac.uk/gwas.
Usage
open_in_gwas_catalog(
identifier,
gwas_catalog_entity = c("study", "variant", "trait", "gene", "region", "publication")
)
Arguments
identifier |
A vector of identifiers. The identifiers can be: study accession identifiers, variant identifiers, EFO trait identifiers, gene symbol names, cytogenetic regions, or PubMed identifiers. |
gwas_catalog_entity |
Either |
Value
Returns TRUE if successful, or FALSE otherwise. But
note that this function is run for its side effect.
Examples
# Open studies in GWAS Web Graphical User Interface
open_in_gwas_catalog(c('GCST000016', 'GCST001115'))
# Open variants
open_in_gwas_catalog(c('rs146992477', 'rs56261590'),
gwas_catalog_entity = 'variant')
# Open EFO traits
open_in_gwas_catalog(c('EFO_0004884', 'EFO_0004343'),
gwas_catalog_entity = 'trait')
# Open genes
open_in_gwas_catalog(c('DPP6', 'MCCC2'),
gwas_catalog_entity = 'gene')
# Open cytogenetic regions
open_in_gwas_catalog(c('2q37.1', '1p36.11'),
gwas_catalog_entity = 'region')
# Open publications
open_in_gwas_catalog(c('25533513', '24376627'),
gwas_catalog_entity = 'publication')
Browse PubMed from PubMed identifiers.
Description
This function launches the web browser and opens a tab for each PubMed citation.
Usage
open_in_pubmed(pubmed_id)
Arguments
pubmed_id |
A PubMed identifier, either a character or an integer vector. |
Value
Returns TRUE if successful. Note however that this
function is run for its side effect.
Examples
open_in_pubmed(c('26301688', '30595370'))
Peels off the _embedded tier from a JSON-list.
Description
This function removes an element named _embedded from the element
content moving all of its contents one level up. If
obj$content$`_embedded` does not exist then returns obj
untouched.
Usage
peel_off_embedded(obj)
Arguments
obj |
A JSON-list. |
Value
A JSON-list.
Creates a platforms table.
Description
Creates a platforms table.
Usage
platforms_tbl(study_id = character(), manufacturer = character())
Arguments
study_id |
GWAS Catalog study accession identifier. |
manufacturer |
Platform manufacturer. |
Value
A tibble whose columns are the named arguments
to the function.
Creates a publications table.
Description
Creates a publications table.
Usage
publications_tbl(
study_id = character(),
pubmed_id = integer(),
publication_date = lubridate::ymd(),
publication = character(),
title = character(),
author_fullname = character(),
author_orcid = character()
)
Arguments
study_id |
GWAS Catalog study accession identifier. |
pubmed_id |
PubMed identifier. |
publication_date |
Publication date (online date if available) formatted
as |
publication |
Abbreviated journal name. |
title |
Publication title. |
author_fullname |
Last name and initials of first author. |
author_orcid |
Author's ORCID iD (Open Researcher and Contributor ID). |
Value
A tibble whose columns are the named arguments
to the function.
Simple rapply version that deals with NULL values.
Description
Like rapply, recursive_apply is a recursive
version lapply but contrary to
rapply, recursive_apply does not ignore
NULL values. Each element of the list which is not itself
a list is replaced by the result of applying fn. If down the line
there are data.frames, then their class is preserved.
Usage
recursive_apply(x, fn)
Arguments
x |
A list (of potentially many nested lists). |
fn |
A function of a single argument. |
Value
A list whose non-list elements have been replaced by the result of
applying fn.
Creates an authors' reported genes table.
Description
Creates an authors' reported genes table.
Usage
reported_genes_tbl(
association_id = character(),
locus_id = integer(),
gene_name = character()
)
Arguments
association_id |
A character vector of association identifiers. |
locus_id |
An integer vector of locus identifiers. |
gene_name |
A character vector of gene symbol according to HUGO Gene Nomenclature (HGNC). |
Value
A tibble whose columns are the named arguments
to the function.
Creates a risk alleles table.
Description
Creates a risk alleles table.
Usage
risk_alleles_tbl(
association_id = character(),
locus_id = integer(),
variant_id = character(),
risk_allele = character(),
risk_frequency = double(),
genome_wide = logical(),
limited_list = logical()
)
Arguments
association_id |
A character vector of association identifiers. |
locus_id |
An integer vector of locus identifiers. |
variant_id |
A character vector of variant identifiers. |
risk_allele |
A character vector of risk or effect allele names. |
risk_frequency |
A numeric vector of the frequency of risk or effect alleles. |
genome_wide |
A logical vector. |
limited_list |
A logical vector. |
Value
A tibble whose columns are the named arguments
to the function.
Convert an S4 object into a list
Description
Convert an S4 object into a list
Usage
s4_to_list(s4_obj)
Arguments
s4_obj |
an S4 object |
Value
A list version of the S4 object.
Setup the environment to skip slow tests
Description
This function sets an environment variable TEST_FAST to 'true' which
is used to flag if a test should be skipped or not. If you place
skip_if_testing_is_fast in a test then it will
check if TEST_FAST is 'true', if it is, then it will skip the next
expectations within a test_that() block.
Please note that this function is to be used in gwasrapidd development.
Usage
set_testing_fast()
Value
Returns NULL. This function should be used for its side
effect.
Setup the environment to still run slow tests
Description
This function sets an environment variable TEST_FAST to 'false' which
is used to flag if a test should be skipped or not. If you place
skip_if_testing_is_fast in a test then it will
check if TEST_FAST is 'true', if it is, then it will skip the next
expectations with a test_that() block. Please note that this function
is to be used in gwasrapidd development.
Usage
set_testing_slow()
Value
Returns NULL. This function should be used for its side
effect.
Set operations on GWAS Catalog objects.
Description
Performs set union, intersection, and (asymmetric!) difference on two objects
of either class studies, associations,
variants, or traits. Note that union()
removes duplicated entities, whereas bind() does
not.
Usage
union(x, y, ...)
intersect(x, y, ...)
setdiff(x, y, ...)
setequal(x, y, ...)
Arguments
x, y |
Objects of either class studies, associations, variants, or traits. |
... |
other arguments passed on to methods. |
Value
An object of the same class as x and y, i.e.,
studies, associations, variants,
or traits.
Examples
#
# union()
#
# Combine studies and remove duplicates
union(studies_ex01, studies_ex02)
# Combine associations and remove duplicates
union(associations_ex01, associations_ex02)
# Combine variants and remove duplicates
union(variants_ex01, variants_ex02)
# Combine traits and remove duplicates
union(traits_ex01, traits_ex02)
#
# intersect()
#
# Intersect common studies
intersect(studies_ex01, studies_ex02)
# Intersect common associations
intersect(associations_ex01, associations_ex02)
# Intersect common variants
intersect(variants_ex01, variants_ex02)
# Intersect common traits
intersect(traits_ex01, traits_ex02)
#
# setdiff()
#
# Remove studies from ex01 that are also present in ex02
setdiff(studies_ex01, studies_ex02)
# Remove associations from ex01 that are also present in ex02
setdiff(associations_ex01, associations_ex02)
# Remove variants from ex01 that are also present in ex02
setdiff(variants_ex01, variants_ex02)
# Remove traits from ex01 that are also present in ex02
setdiff(traits_ex01, traits_ex02)
#
# setequal()
#
# Compare two studies objects
setequal(studies_ex01, studies_ex01)
setequal(studies_ex01, studies_ex02)
# Compare two associations objects
setequal(associations_ex01, associations_ex01)
setequal(associations_ex01, associations_ex02)
# Compare two variants objects
setequal(variants_ex01, variants_ex01)
setequal(variants_ex01, variants_ex02)
# Compare two traits objects
setequal(traits_ex01, traits_ex01)
setequal(traits_ex01, traits_ex02)
Skips a test if TEST_FAST is 'true'
Description
This function checks the value of TEST_FAST, if it is 'true' it skips
the test otherwise it still runs the following expectations. Please note that
this function is to be used in gwasrapidd development.
Usage
skip_if_testing_is_fast()
Value
Returns TRUE if the environment variable 'TEST_FAST' is
set to 'false', or does not return anything but triggers the side
effect of skipping the next test (with testthat).
Constructor for the S4 studies object.
Description
Constructor for the S4 studies object.
Usage
studies(
studies = studies_tbl(),
genotyping_techs = genotyping_techs_tbl(),
platforms = platforms_tbl(),
ancestries = ancestries_tbl(),
ancestral_groups = ancestral_groups_tbl(),
countries_of_origin = countries_tbl(),
countries_of_recruitment = countries_tbl(),
publications = publications_tbl()
)
Arguments
studies |
A |
genotyping_techs |
A |
platforms |
A |
ancestries |
A |
ancestral_groups |
A |
countries_of_origin |
A |
countries_of_recruitment |
A |
publications |
A |
Value
An object of class studies.
An S4 class to represent a set of GWAS Catalog studies
Description
The studies object consists of eight slots, each a table
(tibble), that combined form a relational database of a
subset of GWAS Catalog studies. Each study is an observation (row) in the
studies table — main table. All tables have the column
study_id as primary key.
Slots
studies-
- study_id
GWAS Catalog study accession identifier, e.g.,
"GCST002735".- reported_trait
Phenotypic trait as reported by the authors of the study, e.g.
"Breast cancer".- initial_sample_size
Free text description of the initial cohort sample size.
- replication_sample_size
Free text description of the replication cohort sample size.
- gxe
Whether the study investigates a gene-environment interaction.
- gxg
Whether the study investigates a gene-gene interaction.
- snp_count
Number of variants passing quality control.
- qualifier
Qualifier of number of variants passing quality control.
- imputed
Whether variants were imputed.
- pooled
Whether samples were pooled.
- study_design_comment
Any other relevant study design information.
- full_pvalue_set
Whether full summary statistics are available for this study.
- user_requested
Whether the addition of this study to the GWAS Catalog was requested by a user.
genotyping_techsA
tibblelisting genotyping technologies employed in each study. Columns:- study_id
GWAS Catalog study accession identifier.
- genotyping_technology
Genotyping technology employed, e.g.
"Exome genotyping array","Exome-wide sequencing","Genome-wide genotyping array","Genome-wide sequencing", or"Targeted genotyping array".
platformsA
tibblelisting platforms used per study.- study_id
GWAS Catalog study accession identifier.
- manufacturer
Platform manufacturer, e.g.,
"Affymetrix","Illumina", or"Perlegen".
ancestriesA
tibblelisting ancestry of samples used in each study.- study_id
GWAS Catalog study accession identifier.
- ancestry_id
Ancestry identifier.
- type
Stage of the ancestry sample: either
'initial'or'replication'.- number_of_individuals
Number of individuals comprising this ancestry sample.
ancestral_groupsA
tibblelisting ancestral groups used in each ancestry.- study_id
GWAS Catalog study accession identifier.
- ancestry_id
Ancestry identifier.
- ancestral_group
Genetic ancestry groups present in the sample.
countries_of_originA
tibblelisting countries of origin of samples.- study_id
GWAS Catalog study accession identifier.
- ancestry_id
Ancestry identifier.
- country_name
Country name, according to The United Nations M49 Standard of Geographic Regions.
- major_area
Region name, according to The United Nations M49 Standard of Geographic Regions.
- region
Sub-region name, according to The United Nations M49 Standard of Geographic Regions.
countries_of_recruitmentA
tibblelisting countries of recruitment of samples.- study_id
GWAS Catalog study accession identifier.
- ancestry_id
Ancestry identifier.
- country_name
Country name, according to The United Nations M49 Standard of Geographic Regions.
- major_area
Region name, according to The United Nations M49 Standard of Geographic Regions.
- region
Sub-region name, according to The United Nations M49 Standard of Geographic Regions.
publicationsA
tibblelisting publications associated with each study.- study_id
GWAS Catalog study accession identifier.
- pubmed_id
PubMed identifier.
- publication_date
Publication date (online date if available) formatted as
ymd.- publication
Abbreviated journal name.
- title
Publication title.
- author_fullname
Last name and initials of first author.
- author_orcid
Author's ORCID iD (Open Researcher and Contributor ID).
Drop any NA studies.
Description
This function takes a studies S4 object and removes any study identifiers
that might have been NA. This ensures that there is always a non-NA
study_id value in all tables. This is important as the study_id
is the primary key.
Usage
studies_drop_na(s4_studies)
Arguments
s4_studies |
An object of class studies. |
Value
An object of class studies.
Creates a studies table.
Description
Creates a studies table.
Usage
studies_tbl(
study_id = character(),
reported_trait = character(),
initial_sample_size = character(),
replication_sample_size = character(),
gxe = logical(),
gxg = logical(),
snp_count = integer(),
qualifier = character(),
imputed = logical(),
pooled = logical(),
study_design_comment = character(),
full_pvalue_set = logical(),
user_requested = logical()
)
Arguments
study_id |
GWAS Catalog study accession identifier. |
reported_trait |
Phenotypic trait as reported by the authors of the study. |
initial_sample_size |
Free text description of the initial cohort sample size. |
replication_sample_size |
Free text description of the replication cohort sample size. |
gxe |
Whether the study investigates a gene-environment interaction. |
gxg |
Whether the study investigates a gene-gene interaction. |
snp_count |
Number of variants passing quality control. |
qualifier |
Qualifier of number of variants passing quality control. |
imputed |
Whether variants were imputed. |
pooled |
Whether samples were pooled. |
study_design_comment |
Any other relevant study design information. |
full_pvalue_set |
Whether full summary statistics are available for this study. |
user_requested |
Whether the addition of this study to the GWAS Catalog was requested by a user. |
Value
A tibble whose columns are the named arguments
to the function.
Map a study id to an association id
Description
Map a study accession identifier to an association accession identifier.
Usage
study_to_association(study_id, verbose = FALSE, warnings = TRUE)
Arguments
study_id |
A character vector of study accession identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the study identifier and the second column is the association identifier.
Examples
## Not run:
# Map GWAS study identifiers to association identifiers
study_to_association(c('GCST001084', 'GCST001085'))
## End(Not run)
Map a study id to a EFO trait id
Description
Map a study accession identifier to a EFO trait identifier.
Usage
study_to_trait(study_id, verbose = FALSE, warnings = TRUE)
Arguments
study_id |
A character vector of study accession identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the study identifier and the second column is the EFO identifier.
Examples
## Not run:
# Map GWAS study identifiers to EFO trait identifiers
study_to_trait(c('GCST001084', 'GCST001085'))
## End(Not run)
Map a study id to a variant id
Description
Map a study accession identifier to a variant accession identifier.
Usage
study_to_variant(study_id, verbose = FALSE, warnings = TRUE)
Arguments
study_id |
A character vector of study accession identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the study identifier and the second column is the variant identifier.
Examples
## Not run:
# Map GWAS study identifiers to variant identifiers
study_to_variant(c('GCST001084', 'GCST001085'))
## End(Not run)
Subset an associations object
Description
You can subset associations by identifier or by position using the
`[` operator.
Usage
## S4 method for signature 'associations,missing,missing,missing'
x[i, j, ..., drop = FALSE]
## S4 method for signature 'associations,numeric,missing,missing'
x[i, j, ..., drop = FALSE]
## S4 method for signature 'associations,character,missing,missing'
x[i, j, ..., drop = FALSE]
Arguments
x |
A associations object. |
i |
Position of the identifier or the name of the identifier itself. |
j |
Not used. |
... |
Additional arguments not used here. |
drop |
Not used. |
Value
A associations object.
Examples
# Subset an associations object by identifier
associations_ex01['22505']
# Or by its position in table associations
associations_ex01[2]
# Keep all associations except the second
associations_ex01[-2]
Subset a studies object
Description
You can subset studies by identifier or by position using the
`[` operator.
Usage
## S4 method for signature 'studies,missing,missing,missing'
x[i, j, ..., drop = FALSE]
## S4 method for signature 'studies,numeric,missing,missing'
x[i, j, ..., drop = FALSE]
## S4 method for signature 'studies,character,missing,missing'
x[i, j, ..., drop = FALSE]
Arguments
x |
A studies object. |
i |
Position of the identifier or the name of the identifier itself. |
j |
Not used. |
... |
Additional arguments not used here. |
drop |
Not used. |
Value
A studies object.
Examples
# Subset a studies object by identifier
studies_ex01['GCST001585']
# Or by its position in table studies
studies_ex01[1]
# Keep all studies except the first
studies_ex01[-1]
Subset a traits object
Description
You can subset traits by identifier or by position using the
`[` operator.
Usage
## S4 method for signature 'traits,missing,missing,missing'
x[i, j, ..., drop = FALSE]
## S4 method for signature 'traits,numeric,missing,missing'
x[i, j, ..., drop = FALSE]
## S4 method for signature 'traits,character,missing,missing'
x[i, j, ..., drop = FALSE]
Arguments
x |
A traits object. |
i |
Position of the identifier or the name of the identifier itself. |
j |
Not used. |
... |
Additional arguments not used here. |
drop |
Not used. |
Value
A traits object.
Examples
# Subset a traits object by identifier
traits_ex01['EFO_0004884']
# Or by its position in table traits
traits_ex01[1]
# Keep all traits except the second
traits_ex01[-2]
Subset a variants object
Description
You can subset variants by identifier or by position using the
`[` operator.
Usage
## S4 method for signature 'variants,missing,missing,missing'
x[i, j, ..., drop = FALSE]
## S4 method for signature 'variants,numeric,missing,missing'
x[i, j, ..., drop = FALSE]
## S4 method for signature 'variants,character,missing,missing'
x[i, j, ..., drop = FALSE]
Arguments
x |
A variants object. |
i |
Position of the identifier or the name of the identifier itself. |
j |
Not used. |
... |
Additional arguments not used here. |
drop |
Not used. |
Value
A variants object.
Examples
# Subset a variants object by identifier
variants_ex01['rs4725504']
# Or by its position in table variants
variants_ex01[3]
# Keep all variants except the third
variants_ex01[-3]
Are you sure?
Description
This function asks you interactively for permission to continue or not. You can specify a custom message before the question and also different messages for a both a positive and negative answer.
Usage
sure(
before_question = NULL,
after_saying_no = NULL,
after_saying_yes = NULL,
default_answer = NULL
)
Arguments
before_question |
String with message to be printed before question. |
after_saying_no |
String with message to be printed after answering
|
after_saying_yes |
String with message to be printed after answering
|
default_answer |
String with answer to question, if run in non-interactive mode. |
Details
If you run this function in non-interactive mode, you should pass an
automatic answer to default_answer: 'yes' or 'no'.
Value
A logical indicating if answer was 'yes'/'y'
(TRUE) or otherwise (FALSE).
Map an EFO trait id to an association id
Description
Map an EFO trait id to an association identifier.
Usage
trait_to_association(efo_id, verbose = FALSE, warnings = TRUE)
Arguments
efo_id |
A character vector of EFO trait identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the EFO trait identifier and the second column is the association identifier.
Examples
## Not run:
# Map EFO trait identifiers to association identifiers
trait_to_association(c('EFO_0005108', 'EFO_0005109'))
## End(Not run)
Map an EFO trait id to a study id
Description
Map an EFO trait id to a study accession identifier.
Usage
trait_to_study(efo_id, verbose = FALSE, warnings = TRUE)
Arguments
efo_id |
A character vector of EFO trait identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the EFO trait identifier and the second column is the study identifier.
Examples
## Not run:
# Map EFO trait identifiers to study identifiers
trait_to_study(c('EFO_0005108', 'EFO_0005109'))
## End(Not run)
Map an EFO trait id to a variant id
Description
Map an EFO trait id to a variant identifier.
Usage
trait_to_variant(efo_id, verbose = FALSE, warnings = TRUE)
Arguments
efo_id |
A character vector of EFO trait identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the EFO trait identifier and the second column is the variant identifier.
Examples
## Not run:
# Map EFO trait identifiers to variant identifiers
trait_to_variant('EFO_0005229')
## End(Not run)
Constructor for the S4 traits object.
Description
Constructor for the S4 traits object.
Usage
traits(traits = traits_tbl())
Arguments
traits |
A |
Value
An object of class traits.
An S4 class to represent a set of GWAS Catalog EFO traits.
Description
The traits object consists of one slot only, a table
(tibble) of GWAS Catalog EFO traits. Each EFO trait is
an observation (row) in the traits table — main table.
Slots
traitsA
tibblelisting EFO traits. Columns:
Drop any NA traits.
Description
This function takes a traits S4 object and removes any EFO trait identifiers
that might have been NA. This ensures that there is always a non-NA
efo_id value in all tables. This is important as the efo_id
is the primary key.
Usage
traits_drop_na(s4_traits)
Arguments
s4_traits |
An object of class traits. |
Value
An object of class traits.
Creates a traits tibble
Description
Creates a traits tibble.
Usage
traits_tbl(efo_id = character(), trait = character(), uri = character())
Arguments
efo_id |
A |
trait |
A |
uri |
A |
Value
A tibble whose columns are the named arguments
to the function.
Trim whitespace.
Description
This function removes leading and trailing white space from strings. Note: this function does no checking on input for performance reasons. So make sure the input is really a character vector.
Usage
tws(x)
Arguments
x |
A |
Value
A character vector.
Creates a gene Ensembl identifiers table.
Description
Creates a gene Ensembl identifiers table.
Usage
v_ensembl_ids_tbl(
variant_id = character(),
gene_name = character(),
ensembl_id = character()
)
Arguments
variant_id |
A character vector of variant identifiers. |
gene_name |
A character vector of gene symbols according to HUGO Gene Nomenclature (HGNC). |
ensembl_id |
A character vector of Ensembl identifiers. |
Value
A tibble whose columns are the named arguments
to the function.
Creates a gene Entrez identifiers table.
Description
Creates a gene Entrez identifiers table.
Usage
v_entrez_ids_tbl(
variant_id = character(),
gene_name = character(),
entrez_id = character()
)
Arguments
variant_id |
A character vector of variant identifiers. |
gene_name |
A character vector of gene symbols according to HUGO Gene Nomenclature (HGNC). |
entrez_id |
A character vector of Entrez identifiers. |
Value
A tibble whose columns are the named arguments
to the function.
Extract variant identifiers from strings of the form rs123-G
Description
This function parses strings of the form "rs123-G" and returns
the name of the variant; it uses the regex -([ATCG\?]+)?$.
Usage
variant_name(risk_allele_names)
Arguments
risk_allele_names |
Value
A character vector of variant names.
Map a variant id to an association id
Description
Map a variant identifier to an association identifier.
Usage
variant_to_association(variant_id, verbose = FALSE, warnings = TRUE)
Arguments
variant_id |
A character vector of variant identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the variant identifier and the second column is the association identifier.
Examples
## Not run:
# Map GWAS variant identifiers to association identifiers
variant_to_association(c('rs7904579', 'rs138331350'))
## End(Not run)
Map a variant id to a study id
Description
Map a variant identifier to a study accession identifier.
Usage
variant_to_study(variant_id, verbose = FALSE, warnings = TRUE)
Arguments
variant_id |
A character vector of variant identifiers. |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two identifiers. First column is the variant identifier and the second column is the study identifier.
Examples
## Not run:
# Map GWAS variant identifiers to study identifiers
variant_to_study(c('rs7904579', 'rs138331350'))
## End(Not run)
Map a variant id to an EFO trait
Description
Map a variant identifier to an EFO trait identifier. Variants are first
mapped to association identifiers, and then to EFO traits. Set the option
keep_association_id to TRUE to keep the intermediate mapping,
i.e., the association identifiers.
Usage
variant_to_trait(
variant_id,
keep_association_id = FALSE,
verbose = FALSE,
warnings = TRUE
)
Arguments
variant_id |
A character vector of variant identifiers. |
keep_association_id |
Whether to keep the association identifier
in the final output (default is |
verbose |
Whether the function should be verbose about the different queries or not. |
warnings |
Whether to print warnings. |
Value
A dataframe of two or three identifiers. If
keep_association_id is set to FALSE, the first column is the
variant identifier and the second column is the EFO trait identifier,
otherwise the variable association_id is also included as the second
column.
Examples
## Not run:
# Map GWAS variant identifiers to EFO trait identifiers
variant_to_trait(c('rs7904579', 'rs138331350'))
# Map GWAS variant identifiers to EFO trait identifiers
# but keep the intermediate association identifier
variant_to_trait(c('rs7904579', 'rs138331350'), keep_association_id = TRUE)
## End(Not run)
Constructor for the S4 variants object.
Description
Constructor for the S4 variants object.
Usage
variants(
variants = variants_tbl(),
genomic_contexts = genomic_contexts_tbl(),
ensembl_ids = v_ensembl_ids_tbl(),
entrez_ids = v_entrez_ids_tbl()
)
Arguments
variants |
A |
genomic_contexts |
A |
ensembl_ids |
A |
entrez_ids |
A |
Value
An object of class variants.
An S4 class to represent a set of GWAS Catalog variants
Description
The variants object consists of four slots, each a table
(tibble), that combined form a relational database of a
subset of GWAS Catalog variants. Each variant is an observation (row) in the
variants table — main table. All tables have the column
variant_id as primary key.
Slots
variantsA
tibblelisting variants. Columns:- variant_id
Variant identifier, e.g.,
'rs1333048'.- merged
Whether this SNP has been merged with another SNP in a newer genome build.
- functional_class
Class according to Ensembl's predicted consequences that each variant allele may have on transcripts. See Ensembl Variation - Calculated variant consequences.
- chromosome_name
Chromosome name.
- chromosome_position
Chromosome position.
- chromosome_region
- last_update_date
Last time this variant was updated.
genomic_contextsA
tibblelisting genomic contexts associated with each variant. Columns:- variant_id
Variant identifier.
- gene_name
Gene symbol according to HUGO Gene Nomenclature (HGNC).
- chromosome_name
Chromosome name.
- chromosome_position
Chromosome position.
- distance
Genomic distance between the variant and the gene (in base pairs).
- is_mapped_gene
Whether this is a mapped gene to this variant. A mapped gene is either an overlapping gene with the variant or the two closest genes upstream and downstream of the variant. Moreover, only genes whose mapping source is 'Ensembl' are considered.
- is_closest_gene
Whether this is the closest gene to this variant.
- is_intergenic
Whether this variant is intergenic, i.e, if there is no gene up or downstream within 100kb.
- is_upstream
Whether this variant is upstream of this gene.
- is_downstream
Whether this variant is downstream of this gene.
- source
Gene mapping source, either
EnsemblorNCBI.- mapping_method
Gene mapping method.
ensembl_idsA
tibblelisting gene Ensembl identifiers associated with each genomic context. Columns:- variant_id
Variant identifier.
- gene_name
Gene symbol according to HUGO Gene Nomenclature (HGNC).
- ensembl_id
The Ensembl identifier of an Ensembl gene, see Section Gene annotation in Ensembl for more information.
entrez_idsA
tibblelisting gene Entrez identifiers associated with each genomic context. Columns:- variant_id
Variant identifier.
- gene_name
Gene symbol according to HUGO Gene Nomenclature (HGNC).
- entrez_id
The Entrez identifier of a gene, see ref. doi:10.1093/nar/gkq1237 for more information.
Drop any NA variants.
Description
This function takes a variants S4 object and removes any variant identifiers
that might have been NA. This ensures that there is always a non-NA
variant_id value in all tables. This is important as the
variant_id is the primary key.
Usage
variants_drop_na(s4_variants)
Arguments
s4_variants |
An object of class variants. |
Value
An object of class variants.
Creates a variants table.
Description
Creates a variants table.
Usage
variants_tbl(
variant_id = character(),
merged = integer(),
functional_class = character(),
chromosome_name = character(),
chromosome_position = integer(),
chromosome_region = character(),
last_update_date = lubridate::ymd_hms()
)
Arguments
variant_id |
A character vector of variant identifiers. |
merged |
A logical vector indicating if a SNP has been merged with another SNP in a newer genome build. |
functional_class |
A character vector of functional classes, see
|
chromosome_name |
A character vector of chromosome names. |
chromosome_position |
An integer vector of chromosome positions. |
chromosome_region |
A character vector of cytogenetic regions. |
last_update_date |
A |
Value
A tibble whose columns are the named arguments
to the function.
Export a GWAS Catalog object to xlsx
Description
This function exports a GWAS Catalog object to Microsoft Excel xlsx file. Each table (slot) is saved in its own sheet.
Usage
write_xlsx(x, file = stop("`file` must be specified"))
Arguments
x |
A studies, associations, variants or traits object. |
file |
A file name to write to. |
Value
Although this function is run for its side effect of writing an xlsx file, the path to the exported file is returned.
Examples
# Initial setup
.old_wd <- setwd(tempdir())
# Save an `associations` object, e.g. `associations_ex01`, to xlsx.
write_xlsx(associations_ex01, "associations.xlsx")
# Cleanup
unlink("associations.xlsx")
setwd(.old_wd)